Polylactic Acid Polyols in Urethane Reactive Hot-Melt and Solventborne Adhesives
In addition to a low carbon footprint, polylactic acid brings value to multiple applications through performance attributes such as a high modulus, excellent solvent and grease resistance, and food contact compliance.

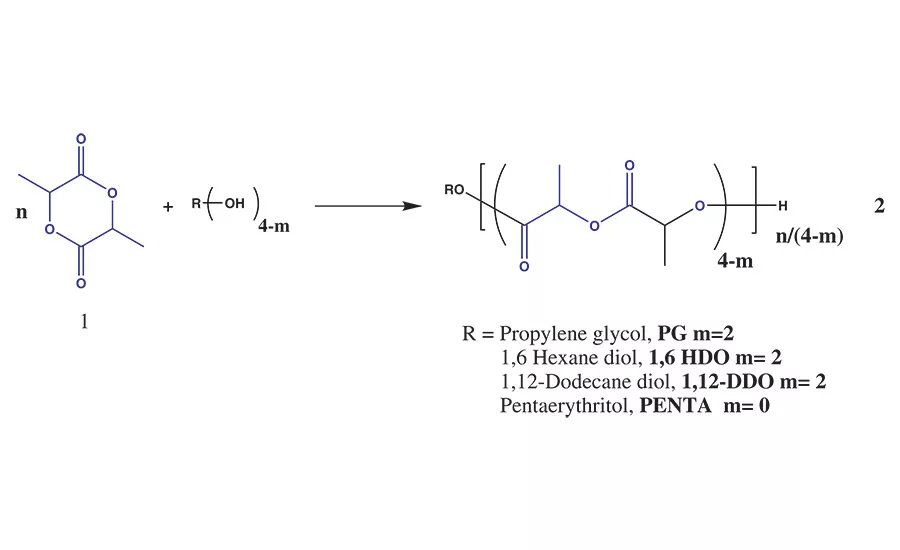
Figure 1. Ring-opening polymerization of lactide with alcohol initiators.
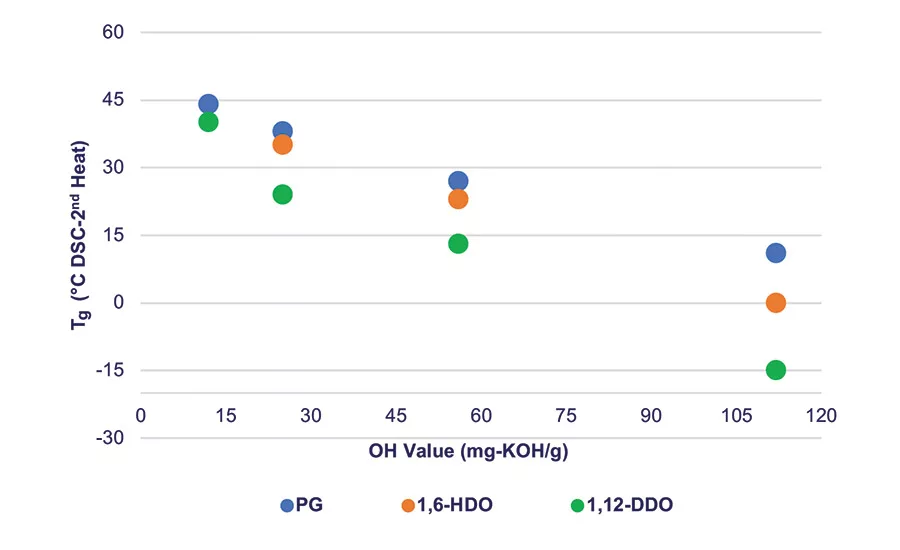
Figure 2. Tg vs. PLA polyol molecular weight for different initiator segments of the PLA polyol.
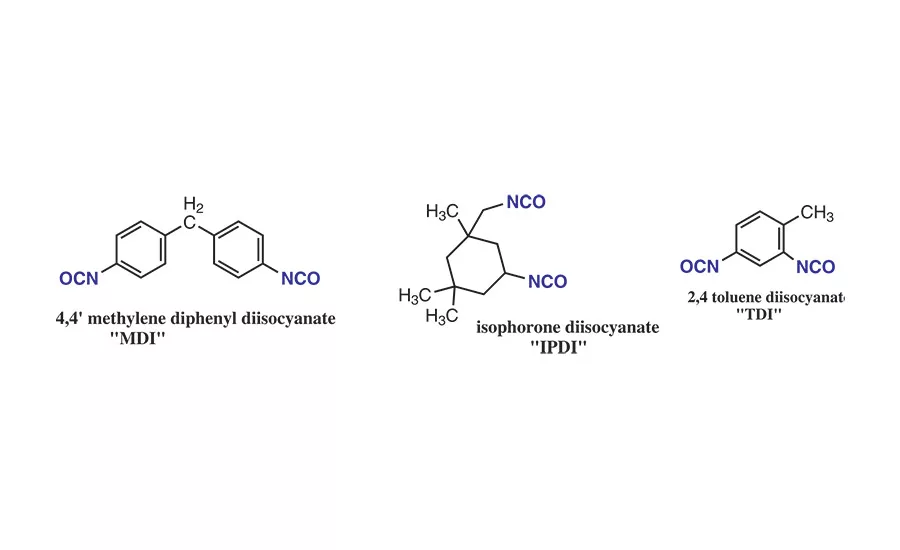
Figure 3. Examples of diisocyanates used in PU-RHMA.
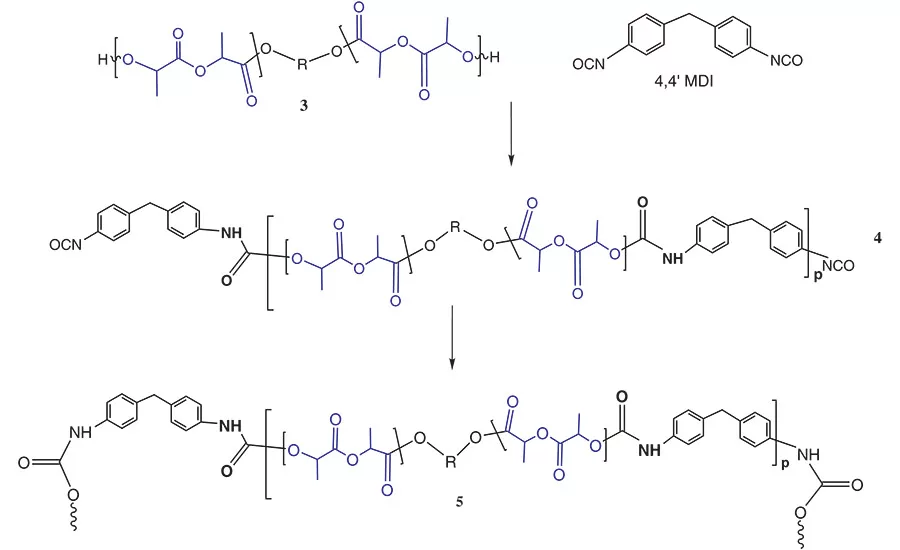
Figure 4. General reaction scheme between PLA polyols and monomeric MDI.
For simplification, this reaction scheme shows the reaction of the PLA NCO prepolymer, 2, with a hydroxyl group to chain extend or crosslink the adhesive via a urethane segment and produce 3. In actuality, this chain extension reaction also occurs between the NCO group and atmospheric and surface water to chain extend and crosslink the polymer via a urea group.
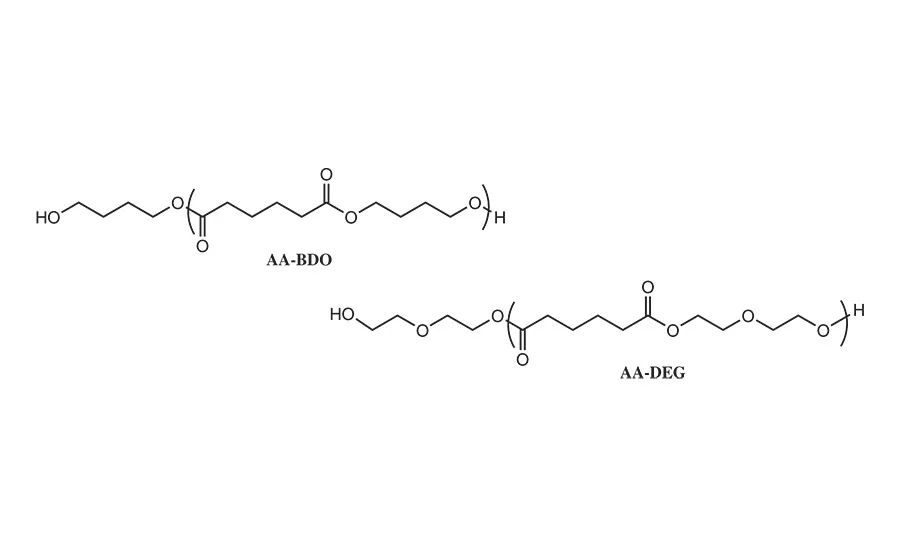
Figure 5. Comparative polyols derived from adipic acid and butane diol and diethylene glycol diol.
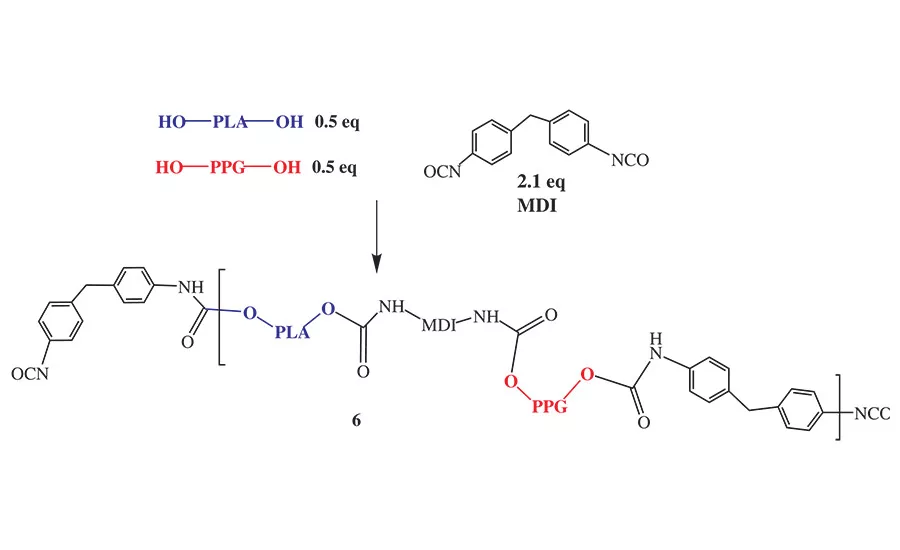
Figure 6. Reaction sequence for NCO prepolymers prepared from blended polyols.
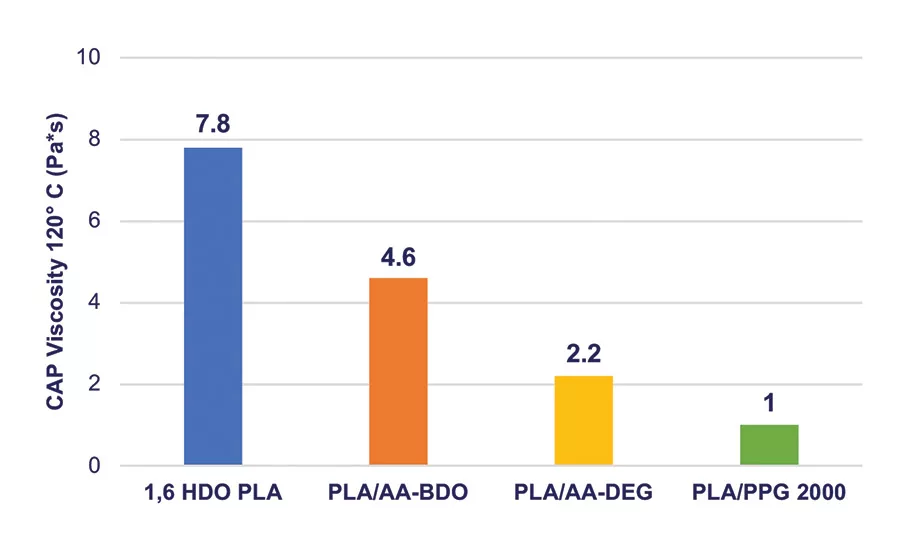
Figure 7. MDI NCO prepolymer viscosity prepared with different polyols. Target NCO content is 3.0-3.5 wt%.
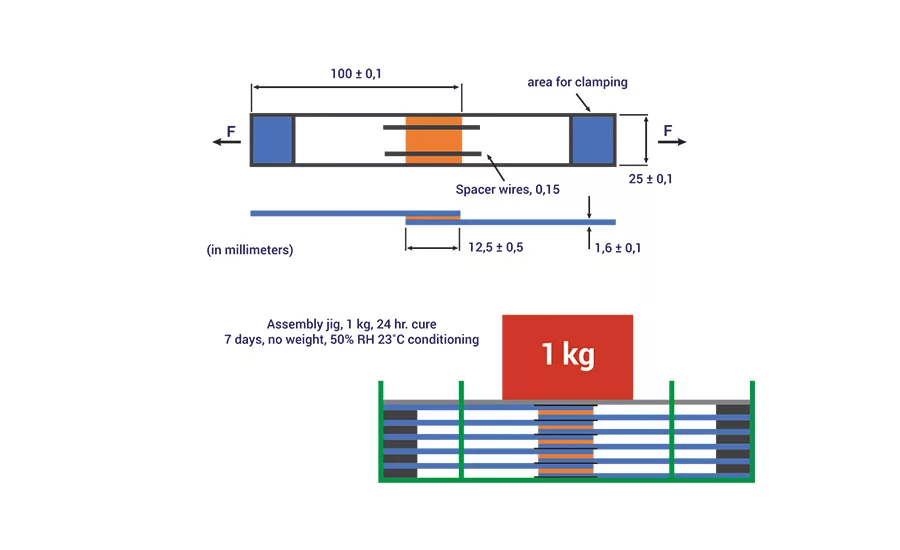
Figure 8. Schematic of the assembly used to prepare the bonded substrates for adhesion testing.
Polylactic acid (PLA) is a well-known bio-based thermoplastic polymer. This versatile and renewable polymer is an example of the commercial success for a bio-based, sustainable polymer with an established and growing technology base.1 The polymer backbone of PLA is based on annually renewable carbon produced via the bacterial or yeast fermentation of sugars currently derived from agricultural feedstocks. The fermentation process produces lactic acid, which is converted to lactide and then polylactic acid via ring-opening polymerization (ROP).
One particular PLA* is used in a variety of applications, including compostable food service ware and packaging, 3D printing filaments, hygiene products, compostable coffee capsules, and nonwovens.2 This PLA brings value to these applications beyond a low carbon footprint through performance attributes such as a high modulus, excellent solvent and grease resistance, and food contact compliance. In addition, it can be industrially composted back to CO2, hummus, and water. The compostable nature of PLA enables this bio-based polymer to be a significant contributor to zero waste and circular economy initiatives focusing on diverting food waste away from landfills.
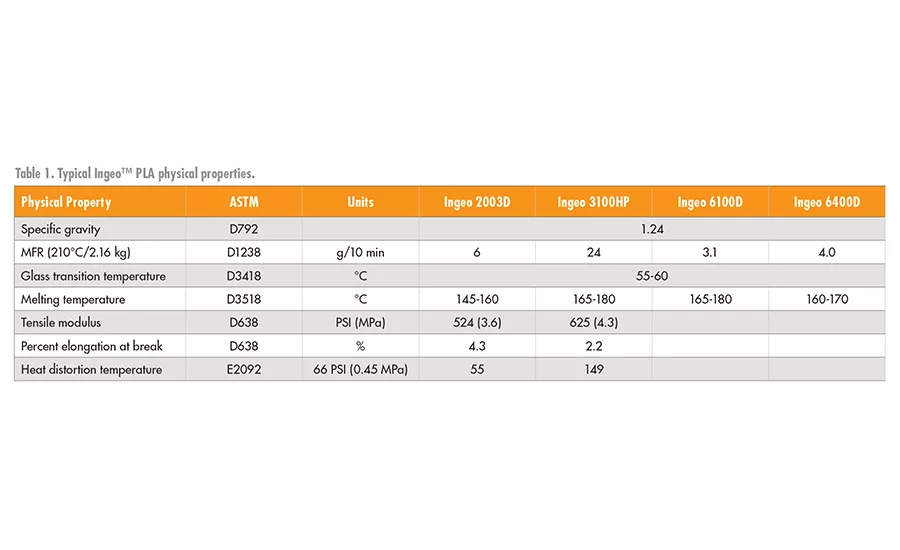
A breadth of physical properties can be achieved by PLA and its compounds that are driven primarily by the control of the PLA molecular weight, degree of crystallinity, melting point, or formulation additives. Some typical physical properties of the PLA are shown in Table 1.3
*Ingeo™ PLA by NatureWorks
Synthesis of PLA Polyols
The polymerization of lactide, 1, can be achieved through the ROP reaction of lactide with an initiating molecule that contains a nucleophile such as amine (-NH) or hydroxyl (-OH) groups to yield high-molecular-weight PLA or PLA polyols, 2, as shown in Figure 1. This reaction is a 100% solids melt polymerization reaction that produces PLA with a well-controlled degree of polymerization, microstructure, and functionality.4,5
When the ROP reaction is initiated by a nucleophile such as a multi-functional alcohol, it is possible to make PLA polyols that are useful reactive intermediates in many coating and adhesive formulations, including urethane chemistry.** By controlling the molecular weight of the PLA oligomer to < 10 kg/mol or a hydroxyl value of > 12 mg-KOH/g for a difunctional polyol, the initiator segment can influence key physical properties of the polyol (e.g., viscosity, glass transition temperature, and solubility) while leaving the terminal secondary hydroxyl groups available for further reactions with functional groups such as isocyanates, anhydrides, caprolactone, glycolide, or other comonomers.
**Vercet™ polyols by NatureWorks
Polyol Characterization
For the sake of consistency, in this article the polyol molecular weight will be referenced by the more commonly used industrial term of hydroxyl value (OH value) of the polyol. This is related to the number-average-molecular weight (Mn) by Equation 1:
Mn = (56,100 * ƒ)/OHV
where 56,100 is the molecular weight of KOH in mg/mol, ƒ is the number of hydroxyl groups per polyol molecule, and OHV is the hydroxyl value in the units of mg-KOH/g-polyol.
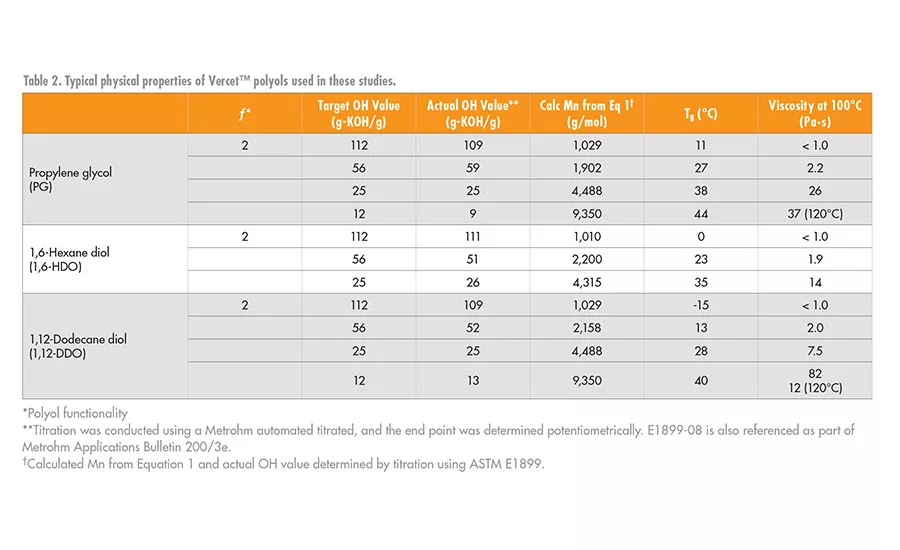
The physical properties of the PLA polyols synthesized with different diol initiators are shown in Table 2. As noted by the changes in the glass transition temperature (Tg) and viscosity values, there is a systematic decrease in the Tg and viscosity values of the polyols as the number of carbons in the initiator increases. Note that at any given hydroxyl value (OH value) between about 12-112 mg-KOH/g, as the length of the initiator segment increases, these values decrease. This shows that the initiator segment influences key physical properties of the PLA polyols and hence can influence the final physical properties of the urethane adhesive.
Figure 2 shows the relationship between Tg, molecular weight, and the polyol initiator. At a OH value of 12 or lower, corresponding to Mn ~ 9,000 g/mol or greater for a difunctional polyol, the Tg of the PLA polyols converges around that of a fully amorphous high-molecular-weight PLA at about 47-50°C. Like the Tg, the viscosity of the polyols also decreases as the chain length of the polyol initiator increases.6
Reactive Hot-Melt Adhesives
Polyurethane reactive hot-melt adhesives (PU-RHMAs) can be prepared from the reaction between the PLA polyols and diisocyanates such 4,4’-methylene diphenyl diisocyanate, isophorone diisocyanate (IPDI), or toluene diisocyanate (TDI). Their general structures are shown in Figure 3.
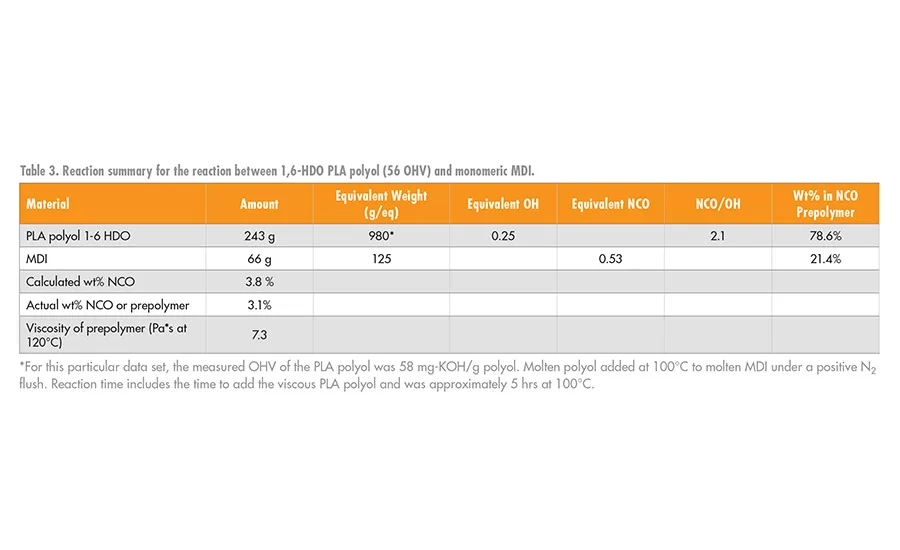
Figure 4 shows the reaction scheme between MDI and a PLA polyol, 3.7 In a PU-RHMA, the reaction stoichiometry between the isocyanate and the polyol is designed to form an isocyanate end-capped oligomer known as a NCO prepolymer, 4. When formulated into an adhesive, the isocyanate groups of the prepolymer further react in the presence of atmospheric moisture or surface hydroxyl groups on the substrate to form additional urethane or urea groups. This additional chemical reaction crosslinks the polymer, 5, to the surface and helps strengthen the adhesive bond that holds the substrates together. PU-RHMAs are particularly useful in wood assembly since these cellulosic substrates have numerous hydroxyl groups and absorbed moisture available to facilitate the final cure of the adhesive to the wooden substrate.
In this study, the synthesis of the NCO prepolymer was formed from the reaction between the PLA polyols of the type shown in Table 2 and the desired isocyanate using standard laboratory glassware, mechanical stirrer, and thermocouple controller. The reactions were conducted under a blanket of nitrogen to minimize unwanted side reactions with water. A tin catalyst was used to improve the reaction rate; a typical reaction preparation is summarized in Table 3. In this initial study, most of the RHMA formulation work focused on the reaction between PLA polyols with 1,6-HDO initiator segments and monomeric MDI. As a comparative system, MDI prepolymers were prepared from the reaction of MDI with butylene diol adipate (AA-BDO) and diethylene glycol adipate (AA-DEG), as shown in Figure 5.
The reaction between MDI and the PLA polyol, 3, was slower owing to the lower reactivity of the secondary hydroxyl groups compared to the reactivity of the primary hydroxyl groups of AA-BDO. However, it was possible to make the desired NCO prepolymer at the target value of 3-3.5 wt% NCO within about 4-6 hrs.
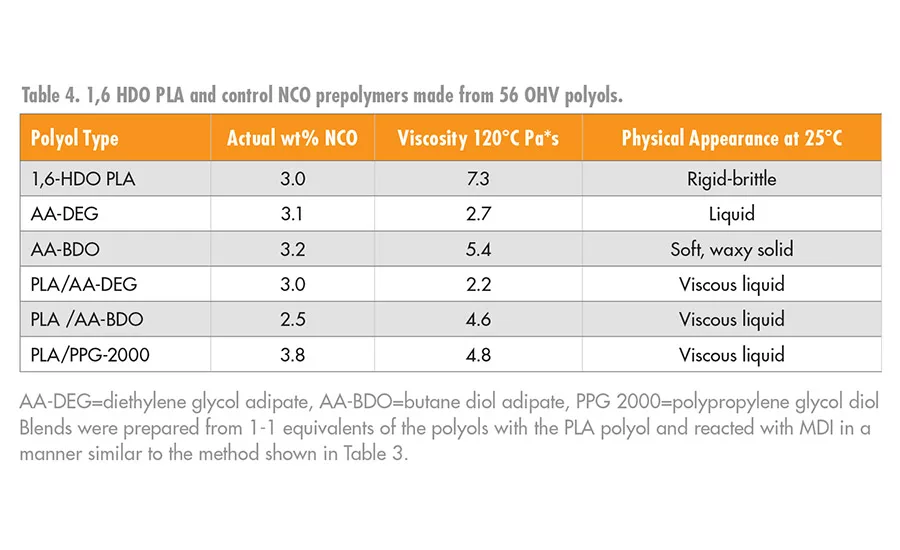
The PLA MDI prepolymers, 4, were higher in viscosity compared to the prepolymers prepared with AA-BDO or AA-DEG. Moreover, the PLA NCO prepolymers tended to be more brittle at room temperature compared to the controls, which made their use in a RHMA challenging. The higher viscosity of the PLA NCO prepolymers was anticipated, given that the PLA polyols have a much higher density of ester groups at a given number of repeat units per OHV than a prepolymer prepared from AA-BDO or AA-DEG. Although high-viscosity adhesives have benefits in non-sag or gap fill applications, they can also be problematic in a RHMA, as it could limit substrate wet out, flow, and open time.
The PLA polyols with a 56 OHV were compatible with AA-DEG, AA-BDO, and PPG diol and formed clear homogenous mixtures in the melt. Therefore, we prepared a series of NCO prepolymers made from equivalent weight mixtures of a PLA polyol and AA-BDO, AA-DEG, or polypropylene glycol diol (PPG diol). The general reaction scheme for the preparation of these copolymers is shown in Figure 6, using PPG diol as the comonomer.
Although Figure 6 suggests the copolymers are in discrete blocks, no detailed characterization of the NCO prepolymer microstructure was made in this study to determine the actual molecular distribution of the polyols in the NCO prepolymer repeat unit. However, since the copolymers were synthesized by first blending the polyols at the desired equivalent ratio and then adding the blend to the molten MDI, it is reasonable to speculate that an NCO prepolymer prepared from a PLA/adipate polyol blend would form some contiguous blocks, while the analogous prepolymer prepared from PLA/PPG polyol blend would be more random. This is a reasonable assumption since the adipate polyols possess primary hydroxyl groups and react faster with MDI than the secondary hydroxyl groups present in the PPG or PLA polyols.
The viscosity and glassy nature of the NCO prepolymers prepared from these blends decreased significantly and had processing characteristics that are more consistent in RHMA applications. Typical data for the homopolymers and copolymers of the NCO intermediates, 4 and 6, are summarized in Table 4, and the viscosity values are shown graphically in Figure 7.
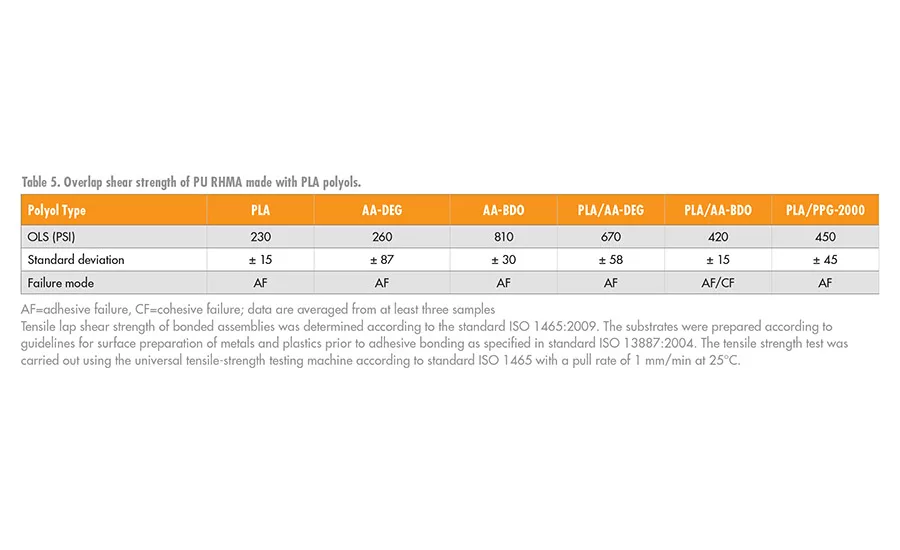
The adhesive performance of the PU-RHMA was evaluated using overlap shear testing according to ISO method 1465. In order to prepare the final RHMA, the NCO prepolymers, 4 or 6, were blended with 1% of glycidyloxypropyl trimethoxy silane adhesion promoter and cured with additional DBTDL catalysts. The adhesives were applied to clean aluminum coupons, clamped with a 1-kg static force, and allowed to cure for 24 hrs. To ensure the adhesives were completely cured before testing, the bonded test specimens were further conditioned without clamping for an additional seven days at 50% relative humidity and 23°C. A schematic of the bond assembly is shown in Figure 8, and OLS adhesive strength data are shown in Table 5.
When PLA was the only polyol used to make the NCO prepolymer, 4, the cured RHMA was a hard, high modulus, brittle material. In contrast, when the NCO prepolymer, 4, was prepared from AA-DEG, the RHMA was soft and flexible. Therefore, as noted in Table 5, both of these RHMA had relatively weak adhesive shear strengths. However, when mixed, these polyols, which form compatible blends, resulted in RHMA matrix that had better adhesion strength then their individual components. This observation suggests that formulation synergies exist between polyols that could be used to tailor the adhesive performance. A formulation DOE would be necessary to find the adhesion optimum that is necessary for the desired application.
Solventborne Polyurethane Adhesives
Solventborne polyurethane adhesives (SB-PUs) have some advantages over 100% solids PU RHMAs. Solventborne adhesives have a low viscosity at room temperature and can be easily applied by numerous coating techniques. Because the adhesive mixture is diluted in a solvent, it is possible to apply very small amounts and form a very thin bond line. The low application viscosity and thin bond line enabled by a SB adhesive are ideal for making heat-seal lamination films that are used in many packaging applications to produce barrier films such as metallized PET films or for overprint varnishes.
Although the solvent is not part of the final adhesive, it often improves the performance of it. During the application, the solvent lowers the surface tension of the polymer and improves the wetting of the adhesive-surface interface. In addition, depending on the nature of the substrate, the solvent can swell the surface of the substrate and improve the penetration of the adhesive into the substrate. This helps to build polymer chain entanglements between the polymer chains and the surface of the substrate, further improving adhesion.
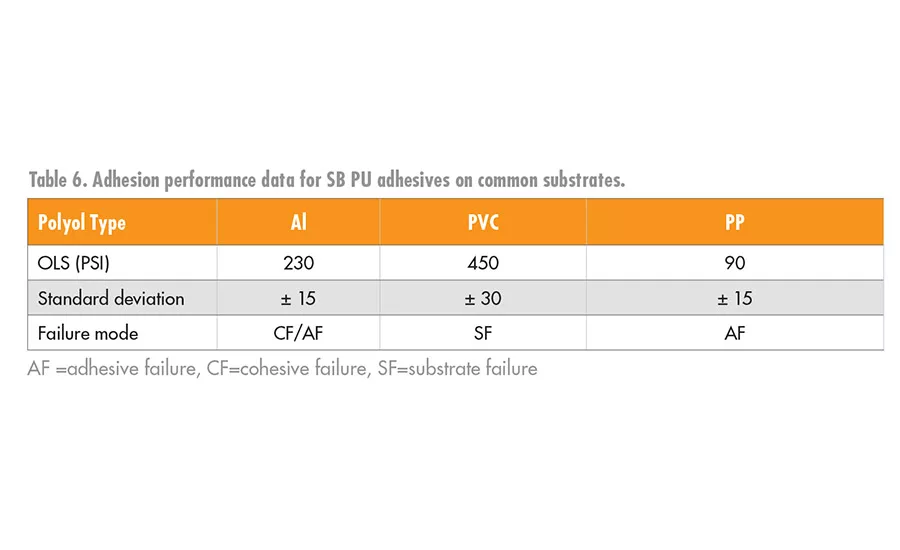
As a result of our findings in our coatings work,8 we screened the use of the PLA polyols in a urethane SB adhesive using HMDI-trimer as the crosslinker with a 112 OHV PLA polyol initiated by propylene glycol. The SB-PU was applied as a 40% solids mixture of the PLA polyol and HMDI trimer (1.1 eq excess of HMDI trimer) to the substrates. The ethyl acetate solvent was allowed to dry off at room temperature; the bond was then formed, clamped, and cured at 80°C for 1 hr. The overlap shear data for the adhesion to Al, PVC, and PP are shown in Table 6.
Developing Bio-Based Adhesive Systems
The polylactic acid polyols discussed provide a sustainable option for polyurethane adhesive applications. The PLA polyol segment in a urethane network provides a viable way to develop bio-based adhesive systems for wood, films, and packaging assembly. Moreover, the properties of PLA could enable adhesives to have biodegradation properties that enable additional end-of-life options for assembled constructions.
Consistent with many other adhesives, the ultimate performance is derived from a formulated system that balances the processing and performance needs of the application. Therefore, the actual formulation needs to be developed within the context and constraints of the application and likely would contain several different components.
For more information, contact the lead author at (952) 562-3308 or bill_coggio@natureworksllc.com, or visit www.natureworksllc.com.
Editor’s note: This article is based on a presentation given at the Adhesive and Sealant Council’s 2019 Annual Spring Conference and Expo. For details regarding the council and its events, visit www.ascouncil.org.
References
-
Polylactic Acid, Synthesis, Structures, Properties, Processing and Applications, Auras, R.; Lim, LT.; Selke, S.E.M.; Tsuji, H., editors, Wiley Scientific, 2010, ISBN978-0-470-29366-9. See also Polylactic Acid PLA Biopolymer Technology and Application, 1st ed., Sin, L.T., editor, Plastic by Design Library, 2012, ISBN 9781437744590.
-
NatureWorks LLC, Product and Application Brochures, www.natureworksllc.com.
-
Ingeo™ Product Brochure and Physical Properties, www.natureworksllc.com.
-
Idage, S.; Idage, B.; Rahman, I.; Sathe., V.; Metkar, S., “Ring Opening Polymerization of Lactide: Kinetics and Modeling,” J. Chem. Eng. Comm., 2019, 206(9) p. 1159.
-
Handbook of Biopolymers and Biodegradable Plastics, Sin, L.T.; Rahmat, A.; Rahman, W.A., Plastic by Design Library, 2013, pp. 55-69, https://doi.org/10.1016/B978-1-4557-2834-3.00003-3.
-
Coggio, W.; Gehrung., M.; Tjosaas, M., “Lactide Polyester Polyols in Polyurethane Coatings,” presented at Coatings Trends and Technology Conference, Chicago, Ill., September 10, 2019.
-
Meier-Westhues, U., Polyurethanes, Vincentz Network Publishers, 2007, Ch. 6, p. 251.
-
Coggio, W., Gehrung, M., Tjosaas, M., “Polylactic Acid Polyols in Urethane Coatings: A Look at the Influence of PLA Polyol Structure on Solvent Borne Urethane Coating Properties,” Paint & Coatings Industry, November 2019, pp. 36-43.
Looking for a reprint of this article?
From high-res PDFs to custom plaques, order your copy today!








