Pulsed Electron Beam Polymerization

Electron beam radiation has been used successfully to crosslink polymers, and it has also been applied to curing applications where ultraviolet (UV) light does not work as well (opaque, etc.).1 It has not been generally applied to polymerization, however, which mostly converts monomers (as opposed to oligomers) and, therefore, requires more time to complete. Most curing applications are used to harden a coating, like a chemical equivalent of drying. If the material under consideration is a pressure-sensitive adhesive (PSA), then meeting the more demanding physical property requirements that depend on polymer structure presents a considerable challenge. To make a high-performance PSA, a high-molecular-weight crosslinked network is essential. Irradiation with an electron beam would be a desirable process for making a PSA because of the advantages of direct initiation: 100%-solids, solventless conversion to a polymer on-web, a product-by-process in one step. However, the difficulty to overcome is the very high dose rate and short residence time at productive speeds, which tend to give low conversion and short chains that may be too tightly networked or that remain as low-molecular-weight residue unincorporated into the gel.2 The main problem with free-radical polymerization under a continuous beam (and other methods) is that neutral reactive species are continuously produced and easily terminate one another. To better understand how this problem may be overcome, and to address all of the issues, the three stages of polymerization must be considered (see Figure 1).
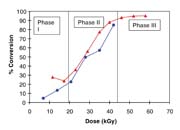
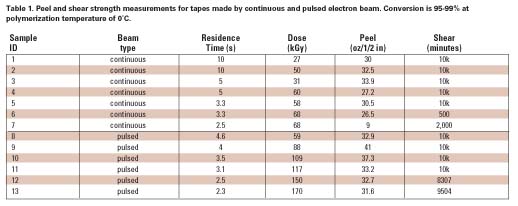
The Three Stages of Polymerization
Phase I
Because initial diffusion rates are high in a low-viscosity system, free radical chemistry is quickly terminated. However, we have learned that low temperature can be advantageously used during this stage to foster chain growth, which is likely the result of a change in shape of the propagating radical in monomer solvent to a ‘balled' structure as illustrated in Figure 2. Thus, it is more difficult to terminate the radical while monomer addition is still relatively easy.3-5 This helps to minimize the initial formation of low-molecular-weight material that later remains unincorporated in the developing gel because it is nonreactive. Even such a small amount of material weakens the bond interface, limiting shear strength (resistance) to only a few hundred minutes. Because e-beams are compact, processing on a temperature controlled drum can be practical, and many commercial units are built in this configuration.
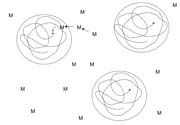
Phase II
At about half-conversion, the polymerization will become diffusion-limited by the large viscosity increase. Both the rate of propagation and termination are slowed, but termination is affected more strongly by orders of magnitude since it involves the reaction of two large polymer radicals. Hence, the rate of conversion increases. At this stage, higher dose rates could be used to finish the polymerization by what are mostly grafting reactions. Ionization now occurs predominantly on the polymer chains, which add monomer to form branched structures. Consequently, conventional e-beam irradiation could be used to finish the polymerization at this stage. Therefore, the need for lower dose rates, low temperature, and longer residence time in order to minimize termination is mostly confined to Phase I.
Phase III
As monomer supply is exhausted, the rate of polymerization slows greatly, and no method can easily provide 100% conversion. Typically, we are left with 2-4% of residual monomers and nonreactive impurities. For many applications, this level of residuals may be acceptable. The removal of these molecules requires evaporation, which would leave a very ‘clean' adhesive suitable for sensitive applications, such as electronics and medical uses, because no contaminates remain. Initiators required by other methods are significant contaminants and more problematic to remove because they are not easily volatilized and can only be removed by extraction. Hence, these high-value specialty applications may justify the development of an e-beam process if the residence time could be shortened further. We were intrigued enough by our earlier results with continuous beam to pursue further improvements that could make e-beam more competitive with other methods.
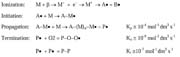
Pulsed Electron Beam for Polymerization
In Phase I, the use of high dose rates becomes self-defeating because the high rate of initiation gives rise to even higher rates of termination (see Figure 3). However, if the beam could be pulsed during an irradiation, no new radicals would be generated during the ‘off' period. During the ‘off' period, surviving radicals would have more time to add monomer before termination could take place by diffusion alone. The effectiveness of the ‘off' time depends on keeping the dose per pulse low enough to provide good separation between the initial radicals. This time period will be extremely short. Unless the pulse rate is high, no advantage would be gained over a continuous beam operated at a low dose rate because the initial advantage of electron track isolation would be quickly lost to diffusion as the system is allowed to become homogeneous. This concept will be further developed in the discussion section of this article.The ability to form short, high-voltage pulses of radiation at fixed intervals is well known and usually necessary to prevent internal arcing. Pulse widths ranging from nanoseconds (pulse radiolysis) to microseconds are achieved using thyratrons. Pulsed low-voltage systems are unavailable commercially, but an experimental device was built for this investigation in which the dose per pulse and the pulse rate could be varied. The requisite equipment flexibility to accomplish this was rendered more easily at low voltages, and low voltage is very compatible with polymerization-on-web (200 keV) where it is necessary to uniformly penetrate only a few mils (10-3 in) of thickness.
To understand how a pulsed beam can affect polymerization, some background is needed to properly frame the discussion. Polymerization that takes place in bulk or in solvent is governed by conventional kinetics based on normal diffusion of species. This type of polymerization is termed ‘homogeneous.' Polymerization that takes place in a phase-separated medium (emulsion, precipitating conditions, or in combination with solid particles of zeolite, catalysts and the like) is governed by special kinetics that account for the compartmentalization of the propagating species. This type of polymerization is termed ‘heterogeneous.' We will describe how the selection of timing (pulse frequency and duration) and spacing (dose per pulse) in a pulsed beam synchronizes with chemical kinetics (diffusion) to compartmentalize a polymerization without the need for phase separation, thus decreasing the residence time required.
Experimental
Materials and Sample Preparation
The monomers were obtained from various chemical companies and used without further purification. A mixture consisting of 89 wt% isooctyl acrylate (IOA) and 11 wt% acrylic acid (AA) was partially polymerized (6% conversion) to form a coatable syrup with a Brookfield viscosity of about 450 centipoises (cP). To this, a crosslinking agent, hexanediol diacrylate (HDODA), was added at an amount of 0.3%. The syrup was coated with a knife coater onto a chemically primed polyethylene teraphthalate (PET) film at a nominal coating thickness of 50 micrometers (2 mils). The coated samples were placed onto a copper plate thermally equilibrated with ice to maintain approximately 0
Results and Discussion
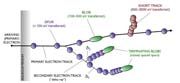
Energy Deposition by Ionizing Radiation is Heterogeneous by NatureEnergy deposited by ionizing radiation results from dislocation of electrons in molecules and is not a homogeneous process. Rather, track-and-spur structures form around each accelerated electron, as generalized in Figure 4. A track traces the path of the primary electron and its secondary electrons through a material, and the spurs are locations along the track where ionization occurs. The dislocated electrons generally return to the ionized molecules to neutralize them and generate an excited state, resulting in bond scission to create a pair of charge-neutral free radicals. These highly reactive fragments then initiate the polymerization. At high electron flux typical of conventional processing, the track-and-spur structures are close enough to overlap, and a homogeneous system results. At low electron flux, there is enough distance between the tracks that a liquid system is initially heterogeneous, but for only a very short period of time before being rendered homogeneous by diffusion. This is known as a low-overlap condition.
Low-Overlap Conditions
In a long series of polymerization experiments, the relationships between dose and degree of conversion were extrapolated to determine the dose required to achieve 97% conversion. The results are plotted in Figure 5 to illustrate the dependency of conversion dose on pulse rate and dose per pulse. In each data series, the pulse duration remains fixed at 1.5 microseconds.
At low pulse rates (<500 Hz), the long time interval between pulses (more than two milliseconds) allows enough time for diffusion to shift the polymerization into the homogeneous mode. This results in a steeper dependence on dose rate because the rate of termination is high. This is exacerbated by high dose per pulse since the distance between the tracks is greatly reduced, which drives polymerization more quickly to the homogeneous mode. At low dose per pulse, the track separation increases and allows the heterogeneous mode to initially dominate the polymerization kinetics, if only for a very short period of time before diffusion renders the system homogeneous.
As the pulse rate increases, the system becomes more dominated by heterogeneous kinetics because progressively less time is allowed for the shift to the homogeneous mode before the system is re-initiated. The progressive flattening of the curves as the dose per pulse decreases and the pulse rate increases shows the dose rate independence that would be expected of a polymerization progressively confined to heterogeneous mode. These conditions are well below the threshold overlap dose,7 at which the system changes from homogeneous to heterogeneous kinetics, and the conversion dose remains low and nearly constant with respect to dose rate.

Some general observations can be made from Figure 5. A polymerization is usually energetically more efficient if the total dose required for conversion is lower because less energy is wasted on termination reactions. Therefore, absolute polymerization efficiency increases as both the pulse rate and dose per pulse decrease. However, where process efficiency is concerned, residence time must be taken into account. Following this consideration, the most desirable operation is at high pulse frequency and low dose per pulse, as will be shown in the next section.
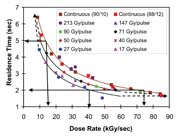
Plotting the results shown in Figure 5 with additional data to express residence time under the beam over a range of conditions (including continuous), we can see the advantage of maintaining the dose per pulse below the threshold overlap. Three operating regimes may be defined, as indicated by the curves in Figure 6: (1) at high dose per pulse (147-483 Gy), pulsed polymerization is no more efficient than continuous polymerization, i.e. the residence times are about the same for both at similar overall dose rates; (2) at a dose per pulse of 50-90 Gy, pulsed polymerization is somewhat more efficient than continuous polymerization; (3) at a dose per pulse of 40 Gy, pulsed polymerization becomes significantly more efficient than continuous polymerization until a leveling occurs, as seen by the similarity between the 27 and 17 Gy data. This is clear evidence that the threshold overlap dose is proximate to 40 Gy, and that track separation is sufficient to provide heterogeneous polymerization below this dose.
As would be expected, the advantage of pulsing is bounded between limits of high and low dose rate. This is shown in Figure 6, in which the curves following pulsed data merge with the continuous case in both extremes. At very low dose rates, around 10 kGy/sec, continuous and pulsed polymerization become indistinguishable, resembling conditions similar to the low dose rates of a gamma source with long residence time. This is the result of sufficiently low flux to allow diffusion to maintain a homogeneous system, regardless of whether new radicals are introduced by pulsing or continuous irradiation. At very high dose rates, around 80 kGy/sec, continuous and pulsed polymerization again become indistinguishable, and both behave as a conventional electron beam with a very short residence time. At these high dose rates, inter-track distances become so small that immediate termination is prevalent regardless of how the new radicals are introduced. Therefore, Figure 6 effectively defines an operating window in which pulsed polymerization can be of benefit.

This method works because, since most of the surviving free radicals generated per pulse are sufficiently far apart, they are momentarily isolated and have a fraction of a millisecond to add monomer before termination reactions begin with neighboring propagating radicals. A kinetic model was made to conceptually explain the polymerization results, showing that the ideal time interval to maintain isolation in this system (EHA) is on the order of a half a millisecond (pulse frequency of 2 kHz), as identified by experiments. Figure 7 shows the results of a kinetic simulation of the propagating free radical concentration. Three conditions were modeled: low, medium and high pulse rates (20 Hz, 100 Hz and 1000 Hz, respectively). The mean dose rates are equal for all three conditions by adjustment of the dose/pulse.
As the time interval between pulses closes, the propagation progressively becomes more confined to the initial part of the curve exhibiting rapid growth with little termination. The peak of the curve represents the point of coalescence of the system, where heterogeneous kinetics begin to transition to homogeneous kinetics. At that point, the advantage of the special kinetics is lost unless the polymerization is effectively terminated as another pulse reinitiates the heterogeneous stage once again. It would appear from examination of Figure 7 that a pulse rate of 2000 pps would further close the interval between pulses and confine the mode of polymerization almost exclusively to the heterogeneous phase, as the experimental results indicate (see Figure 5). As long as the dose per pulse is below the threshold where significant spatial overlap of the electron track-and-spur structures occurs, it is possible to take advantage of heterogeneous (localized) kinetics.8 The rate of polymerization will be proportional to the rate of initiation (RI) rather than the rate of termination (RI ½ ).9
While the polymerization has the appearance of being ‘termination-free,' in reality the system is not termination-free. Termination is being introduced at fixed intervals to re-initiate the polymerization, resulting in nearly the same amount of dose required as for continuous. However, the advantages gained by discrete regulation of the termination reactions are higher molecular weight, dose rate independence and greatly shortened residence time. The electron beam is effectively ‘off' more than 99% of the time during the radiation exposure!
A prototype solid-state unit with variable 4-25 microsecond (µs) pulse duration was built under this program, but is no longer in existence. Currently, a second-generation pulsed beam is being constructed in a joint project between the University of Minnesota, 3M Co. and North Star Power Engineering, a division of Ionatron. The new system uses proprietary technologies developed at North Star Power Engineering that will enable 20µs and shorter pulses at a frequency of 2 kHz, energies of 200 kV, and a peak current of 1 A, while providing excellent reliability.
Among the advantages of a pulsed electron beam is a lower cost than that of a similar direct current (DC) machine. The primary reason for this is that pulsed systems are much less susceptible to vacuum breakdown (arcing) than DC systems, making it easier to insulate the pulsed beam than a DC beam. For this reason, DC machines with a single high-voltage gap above 300 kV have not been built. This is true of electron curtain generators, ion implanters and X-ray tubes. However, single gaps with pulsed voltages of up to 500 kV are routinely used in pulsed systems, such as Klystrons and some research laser systems.
The breakdown properties of oil in pulsed systems also compare favorably to those in DC systems. This allows the high-voltage section of the pulsed electron generator to be smaller and less expensive. In our solid state, 200 kV machine, the dimensions of the pulse transformer are only 20 x 40 x 30 cm.
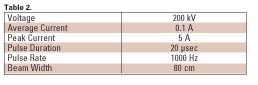
The electron gun used in the prototype machine consisted of a linear thoriated tungsten filament with a beam optics structure designed to give a ~10 cm by 15 cm beam pattern in which the dose was reasonably uniform. Typical parameters for a continuous pilot system are shown in Table 2.
Several similar systems are in use in a material's surface modification. An ion beam produced at 60 kV is used for plasma ion implantation, and these units are robust and reliable.
Conclusion
While free radical chemistry is very fast, it is still possible to intervene and control the course of the chemistry by synchronizing with a pulsed electron beam to introduce space between initiating species and the time intervals necessary for mode-confinement. Pulsed electron beam is a new tool that shows good promise for efficient polymerization on-web to make a product-by-process in a single step. It would likely have application in areas of specialty adhesives, but may also be useful in the development of novel compositions where temperature may be used to control morphology during an ‘instant' polymerization to create novel interpenetrating networks (IPNs) and hydrogels.Acknowledgements
The authors would like to acknowledge the help of fellow 3M employee Roy Schlemmer for conducting some of the many experiments and meticulous measurements necessary to complete this investigation. Benjamin Richter greatly appreciates - and continues to depend on - the guidance of his advisors Professors McCormick and Scriven, as well as support from the Coating Process Fundamentals Program, a program within the Industrial Partnership for Research in Interfacial and Materials Engineering at the University of Minnesota. We would also like to express appreciation to Professors Al-Sheikhly, Silverman and Hongxia Feng of the University of Maryland for their helpful collaboration in extending our understanding of pulsed polymerization and the chemical system currently under study.
For more information on pulsed polymerization, contact Douglas Weiss, Senior Specialist, deweiss@mmm.com.
References
1. Garnett, J. L. "Radiation curing - Twenty Five Years On." Radiat. Phys. Chem. 46, 925-930 (1995).2. Jones, R.A.; Cail, J.I., Stepto, R.F.T.; Ward, I.M. Macromolecules, 33, 7337-7344 (2000).
3. Weiss, D.E., Dunn; D.S. Radiat. Phys. Chem., 65, 281-288 (2002).
4. Wojnarovits, L.; Takacs, E. Radiat. Phys. Chem., 55, 639-644 (1999).
5. Takacs, E.; Dajka, K.; Wojnarovits, L. Radiat. Phys. Chem., 63, 41- 44 (2002).
6. U. S. Patent No. 6,232,365 [WO 0004055], "Low Temperature Electron Beam Polymerization," Douglas Weiss, Bruce Sventek and Thu-Van Tran, April 2000.
7. Kudoh, H.; Celina, M.; Malone, G.M.; Kaye, R.J.; Gillen, K.T.; Clough, R.L. Radiat. Phys. Chem., 48, 555-562 (1996).
8. Henley, E.J.; Johnson, E.R. The Chemistry and Physics of High Energy Reactions, University Press, Washington, D.C., 389 and 396 (1969).
9. Odian, G. Principles of Polymerization, Wiley-Interscience, New York, 208 (1991).
10. Feng, H.; Al-Sheikhly, M.; Silverman, J.; Weiss, D.E.; Neta, P. J. Poly. Sci., Part A: Polymer Chemistry, 41, 196-203 (2002).
Looking for a reprint of this article?
From high-res PDFs to custom plaques, order your copy today!



