Polyesters for Sustainable Adhesive Technologies
Editor’s note: This paper was awarded the Pressure Sensitive Tape Council’s 2011 Carl A. Dahlquist Award for Best Technical Paper.
Melt rheological experiments indicated a correlation between polyester molecular weight and adhesive performance. These studies enabled us to design a new family of segmented copolyesters for their potential in adhesive applications due to controlled sequence distribution and morphological consequences of microphase-separated block copolymers. Incorporating electrostatic interactions into copolymers also affords an avenue for the modification of thermally labile adhesive properties on the molecular level.
We have recently developed a synthetic strategy that allows for melt transesterification of segmented block copolyesters using the sterically hindered and cyclic monomer, 2,2,4,4-tetramethyl-1,3-cyclobutanediol (CBDO). This monomer has also received significant industrial attention as a replacement for bisphenol-A based polycarbonates. High-Tg polyester polyols containing CBDO and 0-5 mol% sulfonation were synthesized using conventional melt transesterification. Further reaction of the hard segment polyols with diesters and diols that comprise the resulting soft block demonstrates a one-pot synthetic method toward a new class of thermoplastic elastomers.
Herein, we describe the influence of ionic interactions on sulfonated segmented copolyesters. Specifically, transesterification resistance studies and melt rheological experiments indicated a correlation between hard segment block content and sequence regularity to the adhesive and mechanical properties of the copolyesters. In addition, atomic force microscopy revealed the surface morphology of the copolyesters. These ion-containing segmented copolyesters permit modification of adhesive properties on both the molecular and morphological level.

Polymer adhesive properties strongly depend on the viscoelastic behavior of the polymers. Viscous behaviors heavily influence the polymer flow properties for processability and product fabrication. However, elastic properties are needed to store and dissipate energy. Elastic properties of adhesives provide the mechanical integrity needed for strong adhesive bonding. The polymer glass-transition temperature also plays a large role in the design of novel polymers for PSA applications. Typical Tg values for room-temperature PSAs fall within the -20 to -60°C range.
In this study, aliphatic low-Tg-based polyesters were synthesized using melt transesterification. The UV stability of aliphatic monomers provides an added advantage that allows these polyester-based adhesives to be suitable for coatings and outdoor applications. Building on the findings of previous research, a new class of segmented block copolyesters has been formulated to improve the mechanical properties of these PSAs.
Recent literature has revealed the importance of introducing secondary interactions into adhesives to improve on the properties of PSAs.1 Melt polymerization of polyesters has afforded polymers with various architectures such as cyclics, copolyesters and hyperbranched polyesters. These architectures have been shown to affect morphology on the nanoscale and tune adhesive properties.2 Due to the transesterification of the ester linkages in the polyester backbone, it is often difficult to achieve segmented polyester. In order to reduce transesterification probability, we use the bulky and rigid cycloaliphatic diol 2,2,4,4-tetramethyl-1,3-cyclobutanediol (CBDO) to produce high-Tg polyester polyols. CBDO has been researched extensively and has been shown to impart high glass-transition temperatures, as well as excellent impact resistance properties.3-4
We have found that these entirely polyester-based block copolymers formed well microphase-separated surface morphologies from atomic force microscopy. Secondary interactions provide physical and mechanical integrity, but, unlike chemical crosslinks, these physical crosslinks are thermally labile (which is important for processing methods).5-7 Block copolymers also allow us to introduce ionic domains into specific segments of the block copolymer to facilitate an understanding of the role of ionic interaction on PSA properties.
Polyesters were made from a combination of diols (diethylene glycol, DEG, or triethylene glycol, TEG) and diesters (dimethyl cyclohexane dicarboxylate, DMCD, or dimethyl adipate, DMAP). The reaction was first purged and degassed three times before heating to 200-220°C over a period of five hours. The reaction temperature was subsequently increased to 275°C, and high vacuum (0.1 mm Hg) was applied to the system. Samples were characterized without further purifications.
Low-Tg polyesters were characterized with 1H NMR, differential scanning calorimetry, THF size exclusion chromatography and thermogravimetric analysis. Adhesive properties were characterized using 180° peel testing, holding power testing, and loop tack and rolling ball tack tests.8 Polymer rheological behavior was investigated using melt rheology on a TA Instrument AR 100 stress-controlled 8 mm parallel plate.

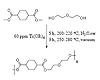
Figure 1 summarizes one class of polyesters that incorporates a cycloaliphatic unit cyclohexane into the polymer backbone. Similarly, Figure 2 outlines the synthesis of polyesters comprising the diester dimethyl adipate, which promotes a noncyclic comparison for DMCD-based polyesters. Diols used for these synthetic schemes were either DEG or TEG. Thus, these monomer choices permit an investigation into the thermal, rheological, and adhesive properties of cyclic- vs. noncyclic-containing polyesters and the influence of an additional ether linkage on the polymer properties.
1H NMR spectroscopy confirmed the structure of these polymers. Table 1 summarizes the polyester polymer composition, as well as molecular weights and Tg values for these polymers. High molecular weights were obtained using melt transesterification methods. These polymers possessed adequately low glass-transition temperatures ranging from -49 to -12°C. The range in the glass-transition temperature can be attributed to the monomer selection, as well as the degree of ether linkages from the choice of diol used.
In general, linear aliphatic polyesters provided lower glass-transition temperatures compared to the cyclohexane-containing polyesters. This property is attributed to the loss of molecular stiffness and molecular bulk when the polyester contained a cyclohexane ring and was not in the linear form. Moreover, the Tg is reduced by 10°C for the DMCD-containing system when the diol is lengthened from DEG to TEG; this is due to the increased flexibility arising from the ether linkage of TEG. These results demonstrate the role of monomer choice on the tailorable properties of these polyester PSAs.
Block copolymers comprising styrene and butadiene or isoprene blocks have been traditionally used in tailoring PSA properties. The drawbacks to these polymer systems are the use of volatile organic solvents during synthesis, as well as the dependence on petroleum-derived monomers. Polyester block copolymers offer a solvent-free approach to synthesize degradable block copolymers for adhesive applications. To build on the work summarized above, an entirely aliphatic-based block copolyester was synthesized. These block copolymers comprised DEG-DMAp soft segment blocks due to the desirable adhesive and thermal properties we described.8 Polyester polyols were synthesized using melt transesterification according to procedures summarized in Figure 3.
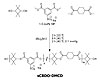
1H NMR spectroscopy confirmed the structure of the sulfonated and unsulfonated polyols systems (see Figure 4). Although chloroform SEC provided appropriate molecular weight results, sulfonated polyols showed aggregation in this solvent. Therefore, molecular weight of the sulfonated system was achieved through 1H NMR spectroscopy. Unsulfonated analogs using dimethyl isophthalate allowed comparison of the glass-transition temperature attributed to ionic molar content.
Using previously determined transesterification study results, the polyester polyols were further used to synthesize segmented copolyesters with DMAp and DEG soft segments using melt transesterification, as shown in Figure 5. 1H NMR confirmed the structure of the segmented copolyesters, and 13C NMR confirmed only two signals for the carbonyl ester corresponding to the carbonyl within the hard segment and within the soft segment.
Dynamic mechanical analysis revealed two thermal transitions corresponding to the soft and hard phases of the segmented copolyesters. The service temperature window ranged from -26 to 120°C, depending on whether the segmented copolyesters were sulfonated or not. The interesting decrease in the rubber plateau modulus prompted further investigation into the segmented copolyesters surface morphology using atomic force microscopy.

These segmented copolyesters were characterized using 1H NMR spectroscopy, TGA, DSC, DMA, tensile tests and AFM. The results suggested the absence of transesterification within the hard segment and microphase separation of the hard and soft blocks. The introduction of ionic groups in the hard segment provides an avenue to tune mechanical and adhesive properties on the molecular level of these copolyesters as potential temperature-sensitive adhesives.

2. Awada, H., Noel, O., Hamieh, T., Kazzi, Y., and Brogly, M., Thin Solid Films, 2011, 519 (11), 3690-3694.
3. Booth, C. J., Kindinger, M., McKenzie, H. R., Handcock, J., Bray, A. V., and Beall, G. W., Polymer, 2006, 47 (18), 6398-6405.
4. Kelsey, D. R., Scardino, B. M., Grebowicz, J. S., and Chuah, H. H., Macromolecules, 2000, 33 (16), 5810-5818.
5. Mather, B. D., Baker, M. B., Beyer, F. L., Green, M. D., Berg, M. A. G., and Long, T. E., Macromolecules, Washington, D.C., 2007, 40 (Copyright (C) 2010 American Chemical Society (ACS). All Rights Reserved.), 4396-4398.
6. Mather, B. D., Baker, M. B., Beyer, F. L., Berg, M. A. G., Green, M. D., and Long, T. E., Macromolecules, Washington, D.C., 2007, 40 (Copyright (C) 2010 American Chemical Society (ACS). All Rights Reserved.), 6834-6845.
7. Saito, T., Mather, B. D., Costanzo, P. J., Beyer, F. L., and Long, T. E., Macromolecules, Washington, D.C., 2008, 41 (Copyright (C) 2010 American Chemical Society (ACS). All Rights Reserved.), 3503-3512.
8. Ozturk, G. I., Pasquale, A. J., and Long, T. E., The Journal of Adhesion, 2010, 86 (4), 395-408.
Figure 1. Synthesis of DMCD-DEG Polymer Using Melt Transesterification Techniques
Abstract
Melt polycondensation synthesis of polyesters presents a solvent- free and environmentally friendly approach of producing pressure-sensitive adhesives (PSAs) due to their inherent biodegradability and low-cost manufacturing process. Low glass-transition temperature (-40°C) all-aliphatic polyester PSAs were synthesized using melt polymerization. Careful selection of diester, diol, and monomer stoichiometry enabled tailoring of the glass-transition temperature (Tg) and adhesive properties as investigated using adhesive testing methods.Melt rheological experiments indicated a correlation between polyester molecular weight and adhesive performance. These studies enabled us to design a new family of segmented copolyesters for their potential in adhesive applications due to controlled sequence distribution and morphological consequences of microphase-separated block copolymers. Incorporating electrostatic interactions into copolymers also affords an avenue for the modification of thermally labile adhesive properties on the molecular level.
We have recently developed a synthetic strategy that allows for melt transesterification of segmented block copolyesters using the sterically hindered and cyclic monomer, 2,2,4,4-tetramethyl-1,3-cyclobutanediol (CBDO). This monomer has also received significant industrial attention as a replacement for bisphenol-A based polycarbonates. High-Tg polyester polyols containing CBDO and 0-5 mol% sulfonation were synthesized using conventional melt transesterification. Further reaction of the hard segment polyols with diesters and diols that comprise the resulting soft block demonstrates a one-pot synthetic method toward a new class of thermoplastic elastomers.
Herein, we describe the influence of ionic interactions on sulfonated segmented copolyesters. Specifically, transesterification resistance studies and melt rheological experiments indicated a correlation between hard segment block content and sequence regularity to the adhesive and mechanical properties of the copolyesters. In addition, atomic force microscopy revealed the surface morphology of the copolyesters. These ion-containing segmented copolyesters permit modification of adhesive properties on both the molecular and morphological level.
Figure 2. Synthesis of DMAP-DEG Polymer Using Melt Transesterification Techniques
Introduction
Acrylic-based block copolymers are typical examples of PSAs that have been traditionally synthesized using volatile organic solvents. Although this class of adhesives serves an important role in commercial products, there remains a large demand for environmentally friendly solvent-free synthetic methods. Researchers have recently synthesized a novel class of low-Tg PSA using melt polymerized polyesters. These polymers harness polyester degradability, as well as solvent-free polymerization techniques, which can essentially lower production costs.Polymer adhesive properties strongly depend on the viscoelastic behavior of the polymers. Viscous behaviors heavily influence the polymer flow properties for processability and product fabrication. However, elastic properties are needed to store and dissipate energy. Elastic properties of adhesives provide the mechanical integrity needed for strong adhesive bonding. The polymer glass-transition temperature also plays a large role in the design of novel polymers for PSA applications. Typical Tg values for room-temperature PSAs fall within the -20 to -60°C range.
In this study, aliphatic low-Tg-based polyesters were synthesized using melt transesterification. The UV stability of aliphatic monomers provides an added advantage that allows these polyester-based adhesives to be suitable for coatings and outdoor applications. Building on the findings of previous research, a new class of segmented block copolyesters has been formulated to improve the mechanical properties of these PSAs.
Recent literature has revealed the importance of introducing secondary interactions into adhesives to improve on the properties of PSAs.1 Melt polymerization of polyesters has afforded polymers with various architectures such as cyclics, copolyesters and hyperbranched polyesters. These architectures have been shown to affect morphology on the nanoscale and tune adhesive properties.2 Due to the transesterification of the ester linkages in the polyester backbone, it is often difficult to achieve segmented polyester. In order to reduce transesterification probability, we use the bulky and rigid cycloaliphatic diol 2,2,4,4-tetramethyl-1,3-cyclobutanediol (CBDO) to produce high-Tg polyester polyols. CBDO has been researched extensively and has been shown to impart high glass-transition temperatures, as well as excellent impact resistance properties.3-4
We have found that these entirely polyester-based block copolymers formed well microphase-separated surface morphologies from atomic force microscopy. Secondary interactions provide physical and mechanical integrity, but, unlike chemical crosslinks, these physical crosslinks are thermally labile (which is important for processing methods).5-7 Block copolymers also allow us to introduce ionic domains into specific segments of the block copolymer to facilitate an understanding of the role of ionic interaction on PSA properties.
Figure 3. Synthesis of Sulfonated Poly (CBDO-DMCD) Using Melt Transesterification
The Experiment
For the experiment materials, polyester monomers were provided by Eastman Chemical Co. and used without further purifications. Titanium catalyst was purchased from Sigma Aldrich and diluted into a 0.01 g/mL 1-butanol solution. The polyesters were synthesized using conventional melt transesterification methods. A 1:1.5 molar ratio of diester to diol and titanium catalyst was charged into a 100-mL round-bottomed flask equipped with a nitrogen inlet, mechanical stirrer and condenser.Polyesters were made from a combination of diols (diethylene glycol, DEG, or triethylene glycol, TEG) and diesters (dimethyl cyclohexane dicarboxylate, DMCD, or dimethyl adipate, DMAP). The reaction was first purged and degassed three times before heating to 200-220°C over a period of five hours. The reaction temperature was subsequently increased to 275°C, and high vacuum (0.1 mm Hg) was applied to the system. Samples were characterized without further purifications.
Low-Tg polyesters were characterized with 1H NMR, differential scanning calorimetry, THF size exclusion chromatography and thermogravimetric analysis. Adhesive properties were characterized using 180° peel testing, holding power testing, and loop tack and rolling ball tack tests.8 Polymer rheological behavior was investigated using melt rheology on a TA Instrument AR 100 stress-controlled 8 mm parallel plate.
Results
Low-Tg polyesters were synthesized in a solvent-free method. The polyester aliphatic backbone contributed to advantages over traditional polystyrene-based PSAs. An aliphatic system enabled coatings or adhesives for outdoor applications due to its increased UV stability over aromatic adhesive systems. In addition, ester linkages enabled degradative properties that may be suitable for time-release adhesives or adhesives for biomedical applications. Thus, this approach allowed the synthesis of a new family of low-Tg polyesters.Figure 1 summarizes one class of polyesters that incorporates a cycloaliphatic unit cyclohexane into the polymer backbone. Similarly, Figure 2 outlines the synthesis of polyesters comprising the diester dimethyl adipate, which promotes a noncyclic comparison for DMCD-based polyesters. Diols used for these synthetic schemes were either DEG or TEG. Thus, these monomer choices permit an investigation into the thermal, rheological, and adhesive properties of cyclic- vs. noncyclic-containing polyesters and the influence of an additional ether linkage on the polymer properties.
1H NMR spectroscopy confirmed the structure of these polymers. Table 1 summarizes the polyester polymer composition, as well as molecular weights and Tg values for these polymers. High molecular weights were obtained using melt transesterification methods. These polymers possessed adequately low glass-transition temperatures ranging from -49 to -12°C. The range in the glass-transition temperature can be attributed to the monomer selection, as well as the degree of ether linkages from the choice of diol used.
In general, linear aliphatic polyesters provided lower glass-transition temperatures compared to the cyclohexane-containing polyesters. This property is attributed to the loss of molecular stiffness and molecular bulk when the polyester contained a cyclohexane ring and was not in the linear form. Moreover, the Tg is reduced by 10°C for the DMCD-containing system when the diol is lengthened from DEG to TEG; this is due to the increased flexibility arising from the ether linkage of TEG. These results demonstrate the role of monomer choice on the tailorable properties of these polyester PSAs.
Block copolymers comprising styrene and butadiene or isoprene blocks have been traditionally used in tailoring PSA properties. The drawbacks to these polymer systems are the use of volatile organic solvents during synthesis, as well as the dependence on petroleum-derived monomers. Polyester block copolymers offer a solvent-free approach to synthesize degradable block copolymers for adhesive applications. To build on the work summarized above, an entirely aliphatic-based block copolyester was synthesized. These block copolymers comprised DEG-DMAp soft segment blocks due to the desirable adhesive and thermal properties we described.8 Polyester polyols were synthesized using melt transesterification according to procedures summarized in Figure 3.
1H NMR spectroscopy confirmed the structure of the sulfonated and unsulfonated polyols systems (see Figure 4). Although chloroform SEC provided appropriate molecular weight results, sulfonated polyols showed aggregation in this solvent. Therefore, molecular weight of the sulfonated system was achieved through 1H NMR spectroscopy. Unsulfonated analogs using dimethyl isophthalate allowed comparison of the glass-transition temperature attributed to ionic molar content.
Using previously determined transesterification study results, the polyester polyols were further used to synthesize segmented copolyesters with DMAp and DEG soft segments using melt transesterification, as shown in Figure 5. 1H NMR confirmed the structure of the segmented copolyesters, and 13C NMR confirmed only two signals for the carbonyl ester corresponding to the carbonyl within the hard segment and within the soft segment.
Dynamic mechanical analysis revealed two thermal transitions corresponding to the soft and hard phases of the segmented copolyesters. The service temperature window ranged from -26 to 120°C, depending on whether the segmented copolyesters were sulfonated or not. The interesting decrease in the rubber plateau modulus prompted further investigation into the segmented copolyesters surface morphology using atomic force microscopy.
Figure 4. 1H NMR of Sulfonated Poly(CBDO-DMCD) High Tg Polyester Polyols in CDCl3
Conclusions
Segmented copolyesters offer an array of desirable attributes, such as tunable mechanical properties and inherent biodegradability through the hydrolytic ester bond. A novel family of sulfonated segmented copolyesters has been developed based on the melt transesterification of poly(CBDO-DMCD) polyols with low-Tg monomers comprising the soft segment.These segmented copolyesters were characterized using 1H NMR spectroscopy, TGA, DSC, DMA, tensile tests and AFM. The results suggested the absence of transesterification within the hard segment and microphase separation of the hard and soft blocks. The introduction of ionic groups in the hard segment provides an avenue to tune mechanical and adhesive properties on the molecular level of these copolyesters as potential temperature-sensitive adhesives.
Figure 5. Melt Transesterification of CBDO-DMCD-DEG-DMAp Segmented Copolyesters Containing Various Sulfonation Molar Content
Acknowledgements
This work is supported in part by the U.S. Army Research Laboratory and the U.S. Army Research Office under the Army Materials Center of Excellence Program, contract W911NF-06-2-0014. The authors would also like to thank Eastman Chemical Co. for its donation of the CBDO and DMCD monomers, and Professor Judy Riffle’s research group for the chloroform GPC analysis.References
1. Lopez, A., Degrandi-Contraires, E., Canetta, E., Creton, C., Keddie, J. L., and Asua, J. M., Langmuir, 2011, 27 (7), 3878-3888.2. Awada, H., Noel, O., Hamieh, T., Kazzi, Y., and Brogly, M., Thin Solid Films, 2011, 519 (11), 3690-3694.
3. Booth, C. J., Kindinger, M., McKenzie, H. R., Handcock, J., Bray, A. V., and Beall, G. W., Polymer, 2006, 47 (18), 6398-6405.
4. Kelsey, D. R., Scardino, B. M., Grebowicz, J. S., and Chuah, H. H., Macromolecules, 2000, 33 (16), 5810-5818.
5. Mather, B. D., Baker, M. B., Beyer, F. L., Green, M. D., Berg, M. A. G., and Long, T. E., Macromolecules, Washington, D.C., 2007, 40 (Copyright (C) 2010 American Chemical Society (ACS). All Rights Reserved.), 4396-4398.
6. Mather, B. D., Baker, M. B., Beyer, F. L., Berg, M. A. G., Green, M. D., and Long, T. E., Macromolecules, Washington, D.C., 2007, 40 (Copyright (C) 2010 American Chemical Society (ACS). All Rights Reserved.), 6834-6845.
7. Saito, T., Mather, B. D., Costanzo, P. J., Beyer, F. L., and Long, T. E., Macromolecules, Washington, D.C., 2008, 41 (Copyright (C) 2010 American Chemical Society (ACS). All Rights Reserved.), 3503-3512.
8. Ozturk, G. I., Pasquale, A. J., and Long, T. E., The Journal of Adhesion, 2010, 86 (4), 395-408.
Looking for a reprint of this article?
From high-res PDFs to custom plaques, order your copy today!






