Semi-Synthetic PSA Generated from Acrylated Biomass

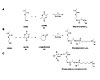
Figure 1. Macromonomers generated with lactide (A) and lactide and ε-caprolactone (B) were used to prepare copolymers of acrylic monomers and macromonomers (C).
Abstract
Consumer goods made from renewable materials, including adhesives and coatings, are required to reduce the use of fossil resources. Research presented here describes an approach to incorporate significant amounts of renewable macromonomers in water-based acrylic polymerization via miniemulsion polymerization systems. The effectiveness of this approach is demonstrated through the generation of several pressure-sensitive adhesive (PSA) latexes produced with 50:50 (mass:mass) of acrylated biomass and 2-ethylhexyl acrylate (EHA). Tested performance properties of adhesive films cast from latexes show values similar to those of commercial products.
Figure 2. Raman spectra of samples L6, L8, L12 and L8C4 (A). The conversion of the acrylate-macromonomer copolymerization in the miniemulsion can be monitored by the disappearance of the double carbon-carbon bond peak at 1640 cm-1 present in both the HEMA-functionalized biomass macromonomers and acrylic monomers, as shown in (B) for the copolymerization of butyl methacrylate with L8 (
Sustainable Solutions
Fossil fuels are the primary source of synthetic chemicals used to produce polymeric consumer goods such as plastics, adhesives, coatings and foams. Most petroleum-based polymers are non-recyclable and non-compostable, and complete their lifecycles as waste. (Plastics and other petroleum-derived materials constitute 20% of municipal solid waste.1) Concerns over the environmental impact of these products, as well as diminishing oil reserves, have driven the pursuit of more sustainable options for our modern needs.Currently, there is considerable interest in biomass derived from woody, herbaceous, and aquatic plants and grasses as a replacement for petroleum-derived feedstocks. The use of biomass for this purpose may lead to the generation of products with closed lifecycles. Furthermore, using non-food energy crops can generate employment, restore fallow lands, and replace agricultural-intensive crops with less intensive crops, resulting in a reduced need for fertilizers and pesticides.2 National security concerns, specifically the need to reduce our dependency on foreign sources for energy and raw materials, offers additional incentives for using biomass in energy and chemical feedstock production. (In 2009, the U.S. imported nearly 60% of its petroleum demand.3)
Discussed here are our efforts to develop PSAs containing high levels of biomass. Pressure-sensitive products have become ubiquitous in our homes and offices. Most of these products contain water-based acrylic PSAs, which are produced mainly from petroleum-based raw materials. More than 300 million dry pounds of acrylic adhesive were sold in the U.S. in 2006.4 A significant portion of this PSA ends its lifecycle in landfills. The introduction of biomass as a substitute for petroleum-based raw materials will make a significant contribution in the development of sustainable products, particularly when these materials can be made biodegradable.
Past efforts to use renewable mass in PSAs have focused mostly on chemicals derived from starch, sugars and vegetable oils. These components are usually not directly compatible with the free radical polymerization used to generate waterborne PSAs and need to be chemically modified to introduce functional groups. Copolymers of acrylic monomers and sugar-derived moieties have been prepared by reacting alkyl polyglycosides with maleic acid anhydride to introduce two functional double bonds that participate in free radical polymerization.5
Although plant-based fatty acids are unsaturated, the double bonds are not reactive enough to directly participate in radical polymerization. Macromonomers from acrylated or hydroperoxidized fatty acids and triglyceride derivatives have been used in miniemulsion polymerizations to prepare waterborne resin coatings. Similarly, acrylated oleate derived from sunflower oil triglycerides were copolymerized with methyl methacrylate and EHA. These copolymer materials demonstrated comparable tack, shear and elasticity compared to commercially available PSAs, although peel values were lower due to the molar mass of the polymers being close to the critical molar mass for physical entanglements.6

Figure 3. Comparison of Loop Tack Values for PSAs Generated with Different MMs
Instead, PSA polymers were generated via miniemulsion polymerization in which the propagation step and chain growth occur in sub-micrometer monomer droplets formed prior to reaction initiation by high shear forces. The absence of mass transport results in faster reaction rates and enables the use of high molar mass components. Monomer droplet stability is achieved through the use of surfactants, but the miscibility of the macromonomer with the other components is critical as well for producing stable miniemulsion precursors and coagulant-free latexes.7,8 Miniemulsions are formed via ultrasonication, which is popular for small laboratory-scale use, but a number of mechanical methods are possible for larger scale production.

Figure 4. Shear as a Function of Added Crosslinker Concentration for Formulations Containing ) and 30 wt% Tackifier Dispersion
Preparation of Lactide-Containing Macromonomers
Similar to plant oil and sugar derivatives, lactide (e.g., derived from corn starch) cannot directly participate in the free radical polymerization used in the preparation of waterborne PSA polymers. The chemical modification of lactide can be achieved by choosing an appropriate initiator for the ring-opening polymerization (ROP). Acrylated polylactic acid (PLA) has been prepared by the ROP of 2-hydroxyethyl methacrylate (HEMA).9-12 This method has been used to prepare copolymers of acrylated PLA macromonomers with n-butyl methacrylate in a miniemulsion process that contained up to 34 wt% biomass.10 Higher biomass incorporation was not possible in this case due to the immiscibility of macromonomers with higher molar mass.Adopting the approach described by Ishimoto et al.,10 acrylated PLA macromonomers (MMs) were prepared with the aim of developing biorenewable PSA polymers by copolymerization of biomass MMs with suitable acrylic monomers (see Figure 1). The addition of a few repeat units of lactic acid in the MM (from 6-12 repeat units) caused a significant increase in the glass-transition temperature (Tg) and a change from a viscous liquid to a waxy solid for 12 repeat units.
Although L6 was miscible with acrylic monomers, a homogeneous mixture of L8 and monomers could only be obtained upon moderate heating and sonication. L12 could not be mixed with the monomer phase and, therefore, could not be used in miniemulsion polymerization. The poor miscibility of the MMs with more than eight repeat units strongly limits the amount of biomass that can be incorporated in the final PSA product and makes processing on larger scales difficult. It was also found to limit the adhesive properties that were achievable via this approach.
Poly(caprolactone) is a liquid polymer at room temperature with a Tg of -60°C. Therefore, a copolymer of repeating units of both ε-caprolactone monomer and semi-crystalline L-lactic acid was expected to have a low Tg, due to the flexibility of the aliphatic ε-caprolactone chain and the suppression of the crystallization of the lactic acid by the statistical incorporation of another monomer. Recently, a process to obtain ε-caprolactone from starch has been described, adding ε-caprolactone to the list of renewable building blocks.13,14 Thus, for our purpose of developing low-Tg biomass-based functional macromonomers, ε-caprolactone is particularly suitable.
As expected, the copolymer MM of lactide and ε-caprolactone (see Figure 1B) are viscous liquids with Tg values ranging from

Preparation of Lactide-Based Biomass PSAs via Miniemulsion
Generated macromonomers were copolymerized with EHA in a miniemulsion polymerization process to produce adhesive polymers (see Figure 1C). Compositions of a small subset of the latexes generated are listed in Table 2.Conventional methods for monitoring the conversion of monomers, such as gravimetry, which rely on the evaporation of the unreacted species, are not suitable for miniemulsion polymerizations due to the presence of the volatile acrylic monomers and the non-volatile MMs. Here, confocal Raman microscopy (CRM) was used to quantitatively determine the fraction of a given functional group by comparing its characteristic peak with a reference peak, as previously described.15 The conversion of the miniemulsion polymerizations was followed by analyzing aliquots of the initial emulsion and during the reaction. Spectra of some of the MMs listed in Table 1 are shown in Figure 2A.
The conversion of the miniemulsion reactions was followed by monitoring the disappearance of the C=C peak at 1642 cm-1, using the peak intensities of the carbonyl groups at 1724 cm-1 (HEMA) and 1768 cm-1 (L and C) as reference peaks. The reaction conversions determined by this method are shown in Figure 2B (p. 32) for the free radical copolymerization of L8 (open circles, ο) and L8C4 (closed squares,·) with an acrylic monomer. The polymerizations were fast, as is typical for miniemulsions, and the reactions proceeded to 90% conversion within the first hour, approaching full conversion after two hours. The solids contents of the latexes were 40 wt%, which is a typical value for macromonomer emulsions without additional polymeric co-surfactants.16

Properties of Lactide-Based Biomass PSAs
Table 3 lists measured values for shear adhesion (ASTM D 6463–99), 180° peel adhesion (ASTM D 903-98) and loop tack (ASTM D 6195-03) for films cast from the biomass-containing latexes listed in Table 2. Also included in Table 2 are two unformulated commercial PSAs: an ultra-removable PSA (PSA1), and a general-purpose permanent PSA (PSA2). In Figure 3, loop tack values are listed for films of biomass-containing copolymers produced with a variety of MMs and for PSA1 and PSA2. It was found that loop tack and peel values are, in general, comparable and are in the range or slightly exceed loop tack of the commercial PSAs used in this study. In addition, the properties of the biomass PSAs can be tuned through the choice of MM.The major challenge with the biomass-containing PSAs is the low shear. The shear adhesion results were affected somewhat by changes in MM composition. An increased crosslink density and the addition of tackifier dispersion had the greatest impact on the shear. For low amounts of tackifying dispersion (< 10 wt%), an increased loop tack was found, as well as a slight decrease in shear. For higher amounts of tackifying dispersions (30 wt%), loop tack values decreased but shear sharply increased (see Table 3).
A decrease in shear is typically observed on the addition of tackifying resin. The strong increase in shear for the biomass-containing PSAs is therefore surprising. Possibly, phase separation between the tackifying resin and the biomass phase of the PSA could play a role in reinforcing the adhesive film. Figure 4 shows an example of the effect of the amount of crosslinker (wt% of ethylene glycol dimethacrylate used during polymerization) for formulations containing no tackifier and 30 wt% of tackifier dispersion. As discussed previously, the shear rises sharply with increasing levels of crosslinking, and this effect is amplified through the addition of tackifier dispersion. The adhesive films containing 30 wt% tackifier are slightly opaque (indicating phase separation), which would restrict their use for clear label applications.
Another strategy to increase shear is the use of “hard monomers” such as styrene in the copolymer. Measured shear values for adhesive polymers containing 20 wt% styrene, 40 wt% EHA and 40 wt% MM (L-L10C6) are about 18 minutes with no added tackifier and 88 minutes with 30 wt% tackifier dispersion. However, this was accompanied by low loop tack values of 0.3 lbf and 0.2 lbf, respectively.
This preliminary work demonstrates the effect of the copolymer composition and formulation with additives on adhesive performance. With strategic design of the copolymer composition and additive addition, it is expected that formulations satisfying shear and loop tack requirements for a variety of applications will be accessible for these biomass-containing PSAs.

Summary and Future Work
An approach was described to incorporate significant amounts of renewable macromonomers in water-based acrylic via miniemulsion polymerization. The effectiveness of this approach is demonstrated through the generation of a series of PSA latexes containing 50 wt% of biomass and 50 wt% EHA. Testing of adhesive films cast from these latexes shows that tack and peel values comparable to commercial PSAs can be achieved for unformulated biomass-containing copolymers. Shear values were enhanced by increasing the concentration of crosslinker reagent added during polymerization reactions and, surprisingly, the addition of tackifier dispersion to the latex. As an example of the possibilities of strategic copolymer design, the strong increase in shear for a copolymer containing styrene as a “hard monomer” was shown.Future work will include a more extensive investigation into the addition of functional monomers in the copolymer and the effect on adhesive performance, as well as an extension of this hybrid copolymer approach to include other renewable materials in PSAs. Semi-synthetic copolymers are currently being developed based on renewable monomers and acrylates that are recycling compatible and fully biodegradable. Initial results indicate that, with the hybrid copolymer approach, the means for producing both recycling compatible and compostable, commercially feasible biomass-based products will be available.
For more information, contact Steve Severtson at sever018@umn.edu.
References
1. Brown, R.C., Biorenewable Resources: Engineering New Products from Agriculture, Iowa State Press, 2003.2. McKendry, P., Biores. Technol., 2002, 83, 37-46.
3. U.S. Energy Information Administration, www.eia.doe.gov, September 4, 2010.
4. “U.S. Industry Study Labels,” Freedonia Group, 2006 and 2011.
5. Bloembergen, S., McLennan, I.J., Narayan, R., U.S. Patent 6,242,593 B1, June 5, 2001.
6. Bunker, S., Staller, C., Willenbacher, N., Wool, R., Int. J. Adhes., 2003, 23, 29.
7. Li, M., Daniels, E.S., Dimonie, V., Sudol, E.D., El-Aasser, M.S., Macromolecules, 2005, 38, 4183-4192.
8. Lu, Y.S., Larock, R.C., Biomacromolecules, 2007, 8, 3108-3114.
9. Huang, S.J., Onyari, J.M, J. ,em>Macromol.Sci., 1996, A33, 571-584.
10. Ishimoto, K., Arimoto, M., Ohara, H., Kobayashi, S., Ishii, M., Morita, K., Yamashita, H., Yabuuchi, N., Biomacromolecules, 2009, 10, 2719-2723.
11. Lim, D.W., Choi, S.H., Park, T.G., Macromol. Rapid Comm., 2000, 21, 464-471.
12. Shinoda, H., Matyjaszewski, K., Macromolecules, 2001, 34, 6243-6248.
13. Arbaoui, A., Redshaw, C., Polym. Chem., 2010, 1, 801-826.
14. Minami, M., Kozaki, S., U.S. Patent 6,559,275, May 6, 2003.
15. Xu, G.Z.H., Dong, J. P., Zhang, J.G., Severtson, S.J., Houtman, C.J., Gwin, L.E., J. Phys. Chem.,, B 2008, 112, 11907-11914.
16. Do Amaral, M., Asua, J.M., J. Polym. Sci. Pol. Chem., 2004, 42, 4222-4227.
Acknowledgement
This work was supported by the Center for Sustainable Polymers at the University of Minnesota through a grant from the Institute for Renewable Energy and the Environment (IREE RL-0009-09). The authors wish to thank Larry Gwin of Franklin International (Columbus, OH) for helpful feedback and Yuxi Zhao (University of Minnesota) for help in characterizing the performance of the generated PSAs.Looking for a reprint of this article?
From high-res PDFs to custom plaques, order your copy today!



